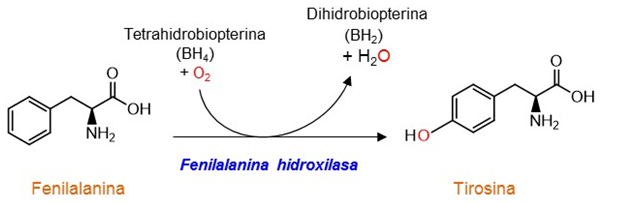
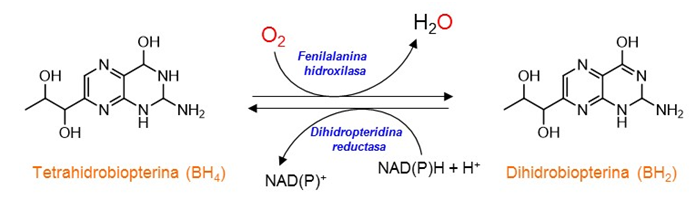
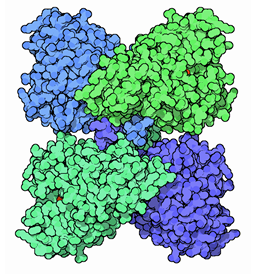
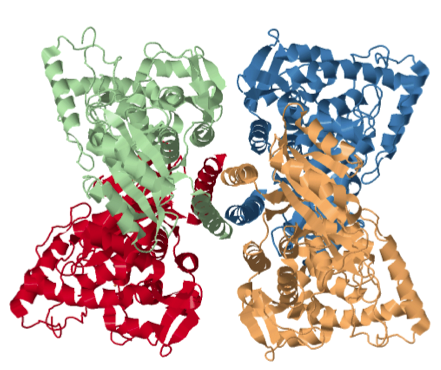
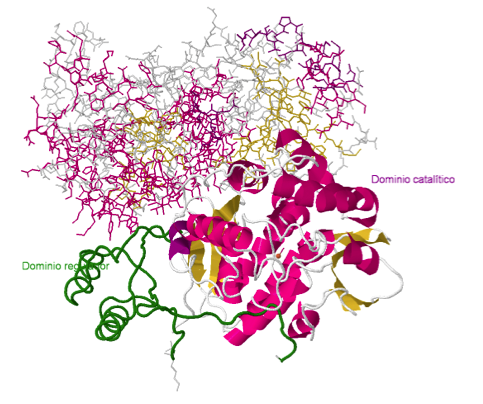
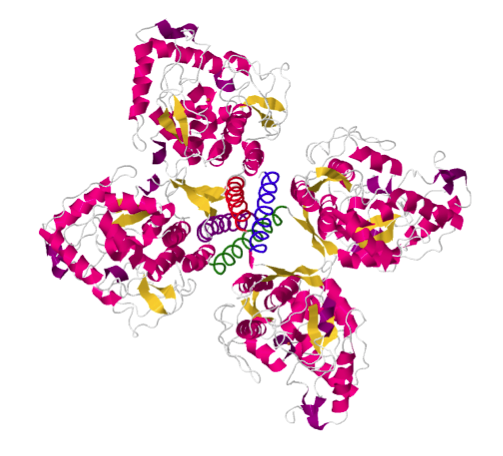
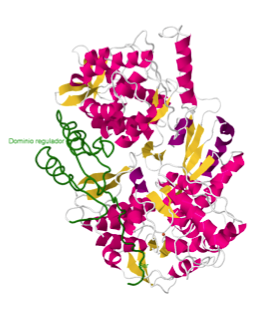
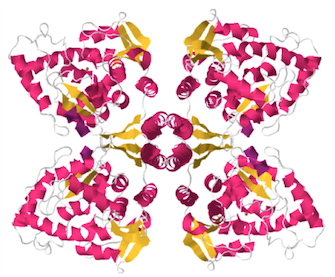
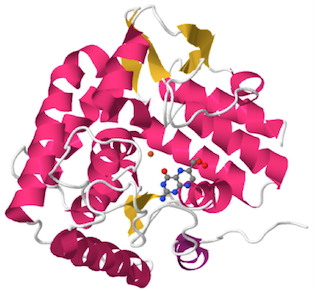
La PAH está íntimamente relacionada con otras enzimas del catabolismo de los aminoácidos aromáticos: la tirosina hidroxilasa que cataliza la hidroxilación de Tyr en la primera reacción de la síntesis de
catecolaminas, y la triptófano hidroxilasa, que cataliza el paso limitante de la síntesis de
serotonina. Las tres enzimas pertenecen a la familia de enzimas denominada “aminoácido aromático hidroxilasas”, monooxigenasas que contienen hierro no hemo, utilizan el cofactor BH4, funcionan como tetrámeros
y presentan la misma organización estructural en tres dominios. El dominio C-terminal está muy conservado en las tres enzimas, pero no así el dominio regulador N-terminal.
La fenilcetonuria
La fenilcetonuria (FCU) es la enfermedad genética más importante relacionada con el metabolismo de los aminoácidos, con una incidencia en la población de 1:10.000. Bioquímicamente se caracteriza por la acumulación
de Phe, y la carencia de Tyr. El exceso de Phe puede metabolizarse a
fenilpiruvato, un compuesto fenilcetónico presente en la orina de personas que padecen fenilcetonuria y del que toma nombre la enfermedad.
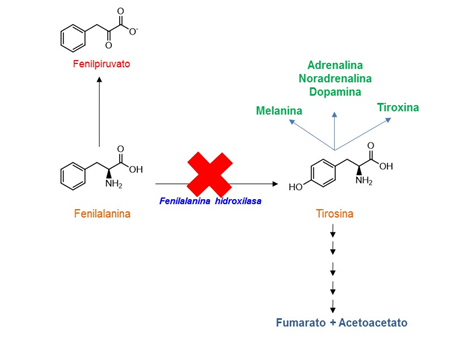
La enfermedad puede deberse a defectos genéticos de la PAH y, más raramente (aproximadamente en el 2 % de los casos), a defectos de las enzimas necesarias para sintetizar la tetrahidrobiopterina (BH4), o para su regeneración
es decir, defectos de la dihidropteridina reductasa (ver la Reacción 2).
Los síntomas de la FCU afectan al sistema nervioso central provocando discapacidad intelectual grave, retraso en el desarrollo, microcefalia y convulsiones, síntomas que en casos no tratados pueden observarse a la edad de un año. Las
causas del retraso mental no se conocen completamente. Se cree que pueden deberse a las altas concentraciones de
fenilpiruvato que sería tóxico para el cerebro. También hay indicios de que la propia Phe sería neurotóxica.
El diagnóstico precoz de la enfermedad es importante porque puede tratarse con medidas dietéticas, como mínimo, hasta que se complete la maduración del cerebro. Por ello, en los recién nacidos se realiza la “prueba del talón”
para la detección temprana de la enfermedad. Mediante esta prueba se detectan los niveles de Phe en sangre de los neonatos con FCU, que pueden llegar a ser 20 veces superiores a los normales.
El tratamiento eficaz de esta enfermedad consiste en la restricción alimentaria de Phe. La Phe se consigue mantener en sangre en niveles próximos a los normales mediante la administración de preparados de aminoácidos sintéticos sin
Phe que se complementan con alimentos naturales como frutas, verduras, cereales, seleccionados por su bajo contenido en Phe. El tratamiento debe comenzar en los primeros 7-10 días de vida para prevenir el deterioro cognitivo. Se
recomienda restringir la Phe de los alimentos durante toda la vida.
La FCU es un trastorno autosómico recesivo, siendo 2 % de la población portadores heterocigóticos. El gen de la PAH se localiza en el cromosoma 12, abarca 90 kb del ADN genómico y contiene 13 exones separados por intrones
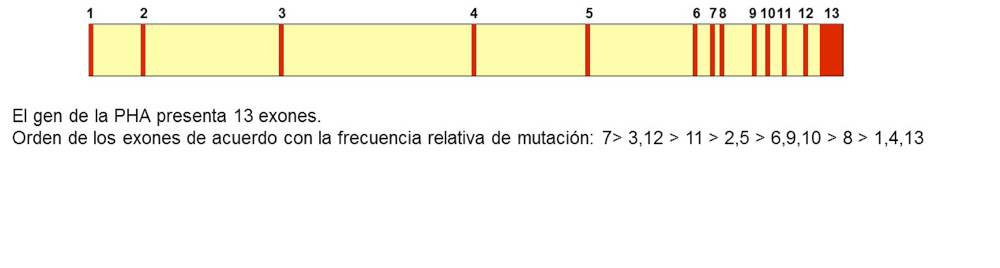
En el gen humano de la PHA se han cartografiado más de 500 mutaciones diferentes. La mayoría de las mutaciones provocan cambios de aminoácidos; también hay mutaciones de corte y empalme, de finalización, silenciosas y deleciones
e inserciones. Muchas de estas mutaciones están recogidas en la base de datos “Phenylalanine Hydroxylase Locus Knowledgebase” http://www.pahdb.mcgill.ca/.
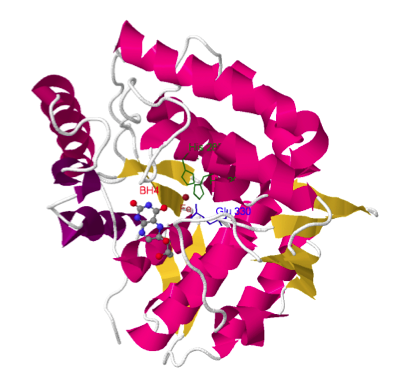
En la imagen siguiente se recogen algunos ejemplos de
mutaciones marcadas en rojo. Estas mutaciones interfieren en la interacción de la enzima con el átomo de
hierro (en naranja) o con el cofactor
BH4 (en verde), reduciendo o eliminando la actividad enzimática.
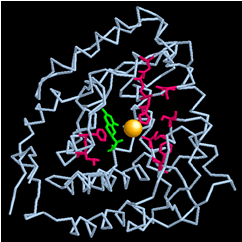 Ejemplos de mutaciones frecuentes de la PHA
Ejemplos de mutaciones frecuentes de la PHA
Muchas de las mutaciones sin sentido provocan defectos en el plegamiento de la proteína. Los monómeros mal plegados tienden a asociarse a través de las regiones hidrofóbicas expuestas y forman grandes agregados proteicos.
Referencias
Dutta, S. & Goodsell D., RCSB PDB Molecule of the Month doi:dx.doi.org/10.2210/rcsb_pdb/mom_2005_1
Erlandsen, H. & Stevens, R.C. (1999) The Structural Basis of Phenylketonuria.
Mol Genet Metab. 68(2):103-125.
Ferrier, D.F. (2014) Lippincott´s Illustrated Reviews: Bioquímica, 6ª Edición, Wolters Kluwer, Barcelona.
Lehninger, A.L.; Nelson, D.L. & Cox, M.M. (2009) Principios de Bioquímica,
5ª ed. Omega, Barcelona.
Ligand Summary page https://www3.rcsb.org/ligand/H4B, información sobre BH4.
McDowall, J. Protein of the Month at the European Bioinformatics
Institute, http://www.ebi.ac.uk/interpro/potm/2005_1/Page1.htm
PDB www.rcsb.org, H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland,
T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne (2000) The Protein Data Bank URL: Nucleic Acids Research, 28: 235-242.
“Phenylalanine Hydroxylase Locus Knowledgebase”.http://www.pahdb.mcgill.ca/