Estructura de la TIROSINA QUINASA c-SRC
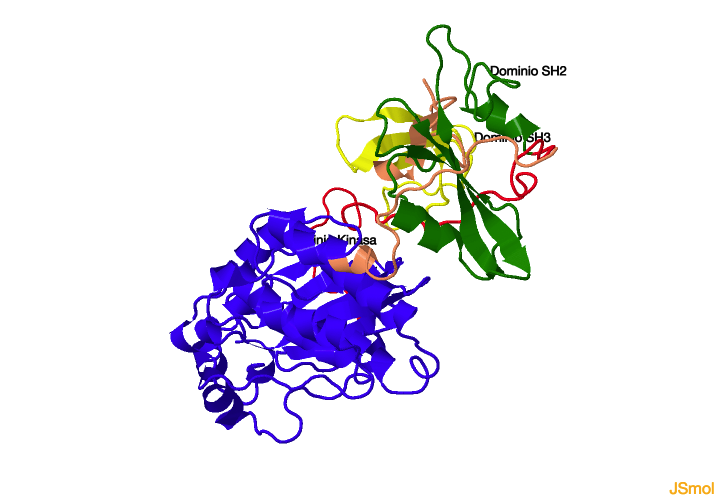
En la siguiente figura se muestra un esquema de las formas activa (izquierda) e inactiva (derecha) de la quinasa c-SRC. En la imagen de la izquierda se indican los distintos dominios de la proteína, desde el extremo N- al C-terminal: un
segmento de anclaje, un dominio SH3, un dominio SH2, una región linker flexible, un dominio quinasa y una cola C-terminal.
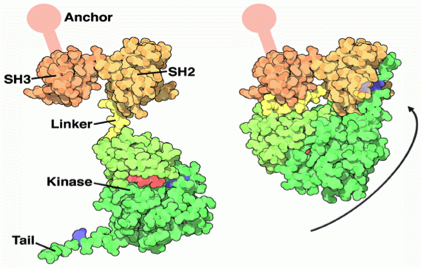 Figura 1: Estructura de la forma activa (izquierda) e inactiva (derecha) de la tirosina quinasa c-SRC
Figura 1: Estructura de la forma activa (izquierda) e inactiva (derecha) de la tirosina quinasa c-SRC
En la conformación activa, el dominio SH3 tiene un pequeño surco que atrapa las cadenas de proteínas sutrato, colocándolas lo suficientemente cerca del dominio quinasa para permitir la adición
de grupos fosfato.
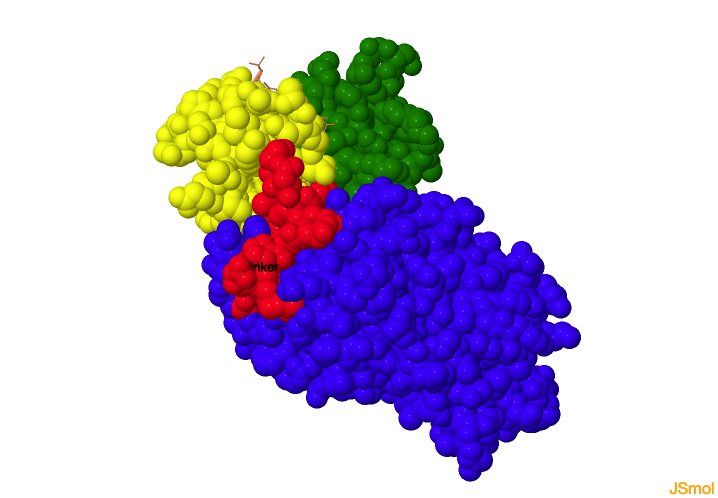
La conformación inactiva de la quinasa es una unión compacta de cuatro dominios. El dominio SH2 y SH3 se extienden respectivamente junto a los dominios mayor y menor de la quinasa. El dominio SH2 forma un puente
salino (combinación de puente de hidrógeno e interacción iónica) con la fosfotirosina 527, y el dominio SH3 se une al dominio quinasa a través de una hélice levógira de poliprolina tipo II. En el siguiente enlace se muestra la
estructura de la proteína c-SRC humana en conformación cerrada. El dominio SH3 se muestra en
amarillo, el dominio SH2 en
verde, el dominio catalítico en
azul, y las zonas linker en
rojo.
Dominios estructurales y funcionales de la tirosina quinasa c-SRC
Catálisis: interacción con el sustrato
Cuando la enzima encuentra en su forma activa y el sustrato se encuentra en el surco que deja el dominio SH3 se produce la fosforilación del sustrato. Esto ocurre gracias a la cesión de un grupo fosfato desde una molécula
de ATP que se encuentra en el dominio quinasa. Como es habitual en las quinasas, el dominio catalítico está formado por dos lóbulos (en
verde y en
azul) enfrentados entre sí sobre todo en la forma inactiva. La conformación del dominio quinasa en la forma inactiva se estabiliza por las interacciones con los dominios SH2 y SH3. La unión del sustrato se produce a nivel del
surco entre ambos lóbulos cuando la quinasa esté en conformación activa. En la conformación cerrada dicho surco está bloqueado por el lazo de activación (en negro en el enlace). En el enlace se indica
la posición de algunos residuos importantes del sitio activo y la región de unión a ATP en color
rojo. La fosforilación/desfosforilación del lazo de activación supone un cambio en la actividad catalítica de la enzima. El lazo de activación no fosforilado adopta una conformación inhibitoria, como se muestra. Gracias a la
fosforilación de la Tyr 416 (marcado en el enlace), el lazo de activación libera el surco existente entre los dos lóbulos de la quinasa, permitiendo la unión con el sustrato. También se ha marcado la posición de la hélice C (en
negro), que ha de desplazarse cuando la quinasa pasa a la conformación activa, lo que permite el reordenamiento de los residuos que participan en la catálisis.
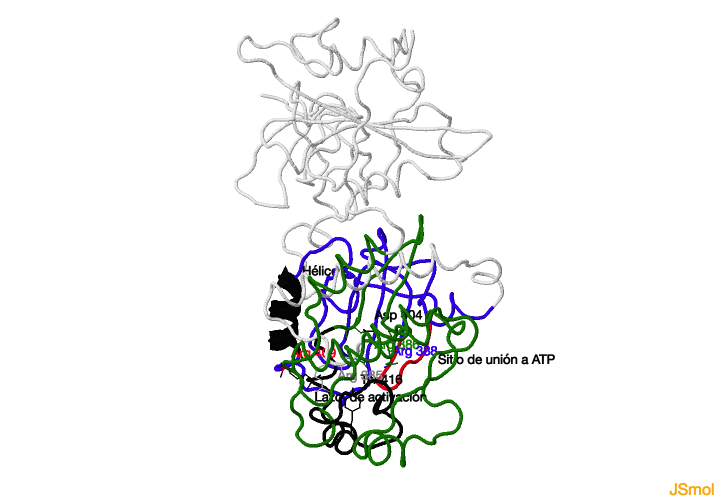
Dominio catalítico de s-SRC
La actividad catalítica completa requiere la separación de los dominios SH3 y SH2 así como el cese de las interacciones moleculares entre ambos, lo cual se consigue con la desfosforilación de Tyr 527 de la cola C-terminal.
Regulación
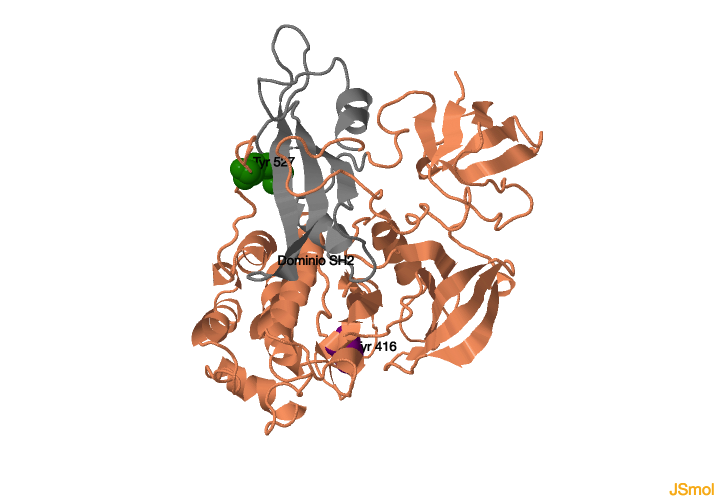
La actividad de la quinasa c-SRC puede regularse de manera positiva o negativa por fosforilación en residuos de tirosina. El plegamiento en la forma inactiva depende de la fosforilación de una tirosina situada en la cola C-terminal
(tirosina 527 en color
verde). Cuando esta tirosina esta fosforilada, se une al dominio SH2 (en color
gris), cerrándose e inactivándose la proteína. Por el contrario, cuando se elimina el fosfato de esta tirosina, se libera la cola y se abre toda la molécula, desbloqueando el sitio de unión de SH3 y permitiendo el acceso
al sitio activo de la quinasa. Por el contrario, la fosforilación de la tirosina 416 del dominio quinasa (en color
morado) provoca la activación de la quinasa.
Residuos de tirosina claves para la regulación de la tirosina quinasa c-SRC
Por tanto, la fosforilación de los residuos de tirosina 416 y 527 tendrán dos efectos contrarios sobre la actividad de Src. La fosforilación de la Tyr 416 media la activación del enzima, mientras que la de la Tyr 527 ejerce el
efecto contrario. Así, la enzima será fosforilada en un residuo o el otro, pero nunca los dos a la vez. En la siguiente figura se resume el modo en que la fosforilación de estos dos residuos influye sobre la actividad de c-SRC.
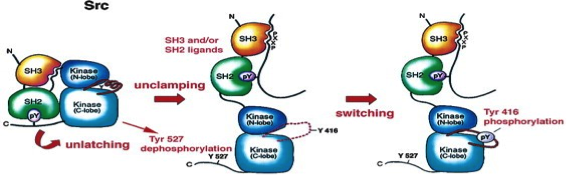 Figura 2: Modelo de activación de Src (regulación covalente)
Figura 2: Modelo de activación de Src (regulación covalente)
La conformación cerrada (inhibitoria) de c-Src está estabilizada por interacciones intramoleculares que se dan entre el dominio quinasa, los dominios SH2 y SH3 y la cola C-terminal fosforilada (Tyr527 fosforilada,
en la figura aparece como pY de phosphorylated tyrosine). Esta fosforilación la lleva a cabo la quinasa del C-terminal de Src (Csk). En este estado, el dominio SH2 se une a la fosfotirosina y a la vez al lóbulo
grande del dominio quinasa. Por otro lado, el dominio SH3 se pone en contacto con el otro lóbulo del dominio quinasa a través de tres residuos de prolina del linker. En este estado, el lazo de activación tiene una conformación
inactiva que hace que el sitio activo de la enzima esté interrumpido mediante el desplazamiento de la hélice C, el cual interfiere con la unión del sustrato y protege a Tyr416 de la fosforilación.
Cuando se unen de manera competitiva ligandos para cada dominio o se desfosforila la Tyr527, SH2 y/o SH3 se desplazan, lo que hace que el dominio quinasa se abra por movimiento de la hélice C y quede expuesta la Tyr416 (susceptible
a fosforilarse).
Por último, la fosforilación de Tyr416 (por un mecanismo de autofosforilación en el dominio quinasa) inicia una reorganización conformacional de todo el lazo de activación, disminuyendo la barrera estérica
para el sustrato y permitiendo que la hélice C regrese al sitio activo. De esta manera, la enzima adquiere la conformación activa.
Papel en la aparición y desarrollo de tumores
El gen src es un gen que fue descrito por primera vez en el sarcoma de Rous, un cáncer que desarrollaban los pollos y que estaba provocado por la infección por un retrovirus (RNA monocaternario) que contiene este gen integrado
en su genoma. Además, se observó que este gen también estaba presente en las células normales de los pollos. Esto llevó a diferenciar dos versiones del gen:
- La versión celular (c-src), que se observaba en los pollos sanos.
- La versión vírica (v-src), que se observaba en los pollos con cáncer.
La diferencia entre ambos es que el primero es un proto-oncogén , imprescindible para la proliferación celular, mientras que el segundo es un oncogén. Los oncogenes virales son genes celulares
(proto-oncogenes) que fueron capturados pos los retrovirus, pasando a ser parte de su genoma. Cuando el virus lo integra, toma únicamente sus exones y es frecuente que en este proceso se produzcan mutaciones puntuales o pérdidas
de fragmentos, alteraciones que derivan en el desarrollo de tumores. Por tanto, se produce una transformación tumoral que hace que se sintetice una proteína tirosina quinasa defectuosa, que está constitutivamente activada y que es independiente de regulación.
 Figura 3: Estructura de las proteínas c-SRC y v-SRC
Figura 3: Estructura de las proteínas c-SRC y v-SRC
En la figura anterior se puede observar que en v-src se produce una pérdida en la cola C-terminal, provocando la pérdida de la tirosina 527. Esto hace que siempre esté en una conformación activa, pues no se puede fosforilar
el residuo que permite adquirir la conformación cerrada. En el siguiente enlace se muestra la estructura de la proteína v-SRC. El dominio SH3 se muestra en
amarillo, el dominio SH2 en
verde, el dominio catalítico en
azul, y las zonas linker en
rojo.
Estructura abierta de v-SRC
Una vez descrito el agente responsable del desarrollo de tumores en pollos, se ha pasado a estudiar con mayor profundidad su homólogo en humanos, c-src. Se ha encontrado que este proto-oncogén está muy implicado en el desarrollo, progresión y metástasis
de numerosos tipos de cáncer en humanos (colon, mama, páncreas, cerebro) y se ha observado que los niveles de esta proteína se encuentran muy elevados en comparación
con tejidos sanos. Además, también se ha mostrado un incremento en su actividad específica. Sin embargo, no hay evidencias de que Src sea un oncogén humano (es decir, no está demostrado que esté implicado
en la génesis de tumores en humanos) y todavía no se han descrito los mecanismos de esta activación. Se cree que puede estar relacionado con mutaciones (que, por ejemplo, reduzcan la actividad del regulador negativo
Csk o que aumenten la actividad de fosfatasas que actúan como reguladores positivos). Una mayor actividad de Src en tumores también podría deberse a la interacción con receptores de tirosina quinasa como EGFR (receptor
del factor de crecimiento epidérmico), PDGFR (receptor del factores de crecimiento de plaquetas), FGFR (receptor del factor de crecimiento de fibroblastos), CSF-1R (receptor del factor estimulante de colonias 1), HER2
y c-Met (receptor del factor de crecimiento de hepatocitos).
Respecto a su papel en la expansión tumoral, los sustratos fosforilados de c-Src alteran la transducción de señales y afectan, por tanto, a eventos de transcripción, lo cual resulta en: alteraciones en la adhesión normal, angiogénesis (por la inducción
de VGF), crecimiento tumoral (por la inducción de Bcl-x1 mediada por Stat3, activado por Src), ciclo celular (por ciclina D1 y c-Myc, inducidas por Stat3) y apoptosis. Se ha observado que c-Src causa también un aumento
de la cantidad de receptores del IGF y, por tanto, mayor crecimiento dependiente de IGF, e incluso la relocalización de una proteína c-Src al núcleo, donde podría influir en la regulación génica, tras el aumento de
su actividad.
Es ahora cuando se están empezando a conocer las múltiples vías en las que participa Src, lo que llevará a un mejor entendimiento de cómo los diferentes sustratos interaccionan con c-Src y llevan a la transformación y progresión
tumoral maligna. Una vez comprendido esto, se puede pasar a estudiar moléculas que tengan como blanco c-Src para poder utilizarlas como terapias contra el cáncer.
Guion elaborado por Blanca Ayuso Íñigo, Yanira González Rodríguez, María Hierro Soto, Sandra Isidro Jorge y Enrique Javato Flores, alumnos de Química e Ingeniería de Proteínas, 3er curso del Grado en Biotecnología, curso
2017/18.
Referencias:
Branden, C. I. (1999). Introduction to protein structure. Garland Science.
Soto Cruz, I. (2008). Proteínas cinasas de la familia Src en el desarrollo del cáncer. Vertientes Revista Especializada en Ciencias de la Salud, 11(1-2), 3-9.
Irby, R. B., & Yeatman, T. J. (2000). Role of Src expression and activation in human cancer. Oncogene, 19(49), 5636.
Roskoski, R. (2004). Src protein–tyrosine kinase structure and regulation. Biochemical and biophysical research communications, 324(4), 1155-1164.
Xu W, Harrison SC, Eck MJ. (1997). Three-dimensional structure of the tyrosine kinase c-Src. Nature, 385(6617):595-602.
Xu, W., Doshi, A., Lei, M., Eck, M. J., & Harrison, S. C. (1999). Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Molecular cell, 3(5), 629-638.